HSP70: Inhibitors
Hsp70 Inhibitors
ATP analogs
Desoxyspergualin / dihydropyrimidines
Flavonoids
Imidazoles
Methylene blue
Peptides
Phenylethylsulfonamide
Rhodocyanines
Sulfoglycolipids
The inhibition of HSP70s has emerged as a promising anticancer strategy in preclinical and clinical trials. Targeting membrane-bound Hsp70-1 by the mouse monoclonal antibody cmHsp70.1 which specifically recognizes a C-terminal epitope of Hsp70-1 might be a promising approach as it selectively induces antibody-dependent toxicity of human tumor cells xenografted in mice 18. In contrast to other commercially available Hsp70 antibodies, cmHsp70.1 mAb uniquely recognizes the membrane form of Hsp70-1 on viable tumor cells 253.
Only a few studies identified chemical compounds that have the capacity to directly modulate the activity of HSP70s. Nevertheless, HSP70 inhibitors have the potential for use in single-agent or combinatorial therapies in order to supplement the hitherto existing conventional chemotherapeutical approaches and molecularly targeted drugs. The most relevant HSP70 inhibitors can be grouped in different categories 92, 254, 255.
ATP analogs
The ATP analog Ver-155008 binds to the NBD of both, Hsp70-8 (Hsc70) and Hsp70-1 (HspA1), and thereby acts as an ATP-competitive inhibitor preventing allosteric control between NBD and SBD 256. VER-155008 was reported to inhibit proliferation of HCT116 human colorectal cancer cells with concomitant reduction in cellular levels of Raf-1 and Her-2 257. VER-155008 also inhibited proliferation of BT474 human breast cancer cells, induced Hsp90 client protein degradation and caspase-3/7 dependent apoptosis in these cells 258. Unfortunately, the bioavailability of VER-155008 is low because it is rapidly degraded in vivo and its level in tumor tissues never reaches the predicted pharmacologically relevant level.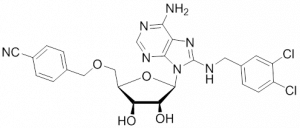
Desoxyspergualin / dihydropyrimidines
15-deoxyspergualin, a synthetic derivative of spergualin from Bacillus spec., was about the first compound known to modulate the ATPase activity of Hsp70-1 and showed significant antitumor activity against L1210 leukemia cells in mice 259. Unfortunately, in a phase II trial 15-deoxyspergualin did not have significant activity against metastatic breast cancer resistant to front-line chemotherapy 260. The dihydropyrimidine NSC 630668-R/1 was identified to inhibit yeast Hsp70-8 (Hsc70) activity and to block Hsp70-8-mediated protein translocation in vitro 261. MAL3-101 represents a second-generation derivative of dihydropyrimidines with blocking Hsp70-1 ATPase activity 261. MAL3-101 was found to exhibit antimyeloma effects on multiple myloma cell lines in vitro and in vivo in a xenograft plasmacytoma model, as well as on primary tumor cells and bone marrow endothelial cells from myeloma patients 262.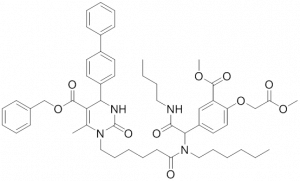
Flavonoids
The catechin (-)-epigallocatechin-3-gallate (EGCG) is the most abundant and powerful catechin in cancer prevention and treatment 263. Pleiotropic effects of EGCG include antioxidant activities, cell signaling modulation, apoptosis induction, cell cycle arrest as well as inhibition of matrix metalloproteinases (MMPs), urokinase-plasminogen activator, telomerase, DNA methyltransferase, and proteasome 264. In a recent study it became apparent that EGCG directly interacts with the ABD of Hsp70-5 (Grp78) thereby inhibiting its ATPase activity 265. EGCG also enhanced etoposide-induced apoptosis in cancer cells by interfering with the antiapoptotic Hsp70-5/caspase-7 complex and suppressed the transformed phenotype after etoposide treatment. These results clearly indicate the inhibitory effect of EGCG on antiapoptotic Hsp70-5 which plays a critical role in the development of drug resistance. The flavonol myricetin has been shown to directly inhibit Hsp70 activity and facilitate chaperone-mediated Tau turnover in Tau-overexpressing HeLa cells 266. The microtubule associated protein Tau plays a central role in neurodegenerative diseases including Alzheimer’s disease that are characterized by the intracellular accumulation of Tau into tangles as a consequence of the accumulation of amyloid plaques composed of Aβ peptide 267. The data given by Jinwal and co-workers clarify the roles of endogenous Hsp70 in Tau processing and suggest an unexpected avenue for therapeutic intervention.
Imidazoles
In 2008, the group of Injae Shin reported on the ability of apoptozole, a small molecule capable of inducing apoptosis, to bind to human Hsp70-1 and Hsp70-8 and thereby to induce apoptosis in human embryonic carcinoma and lung cancer cells 268. Apoptozole inhibits the ATPase activity of Hsp70-8 and presumably of Hsp70-1 by binding to the ATP-binding pocket within its ATPase domain 269.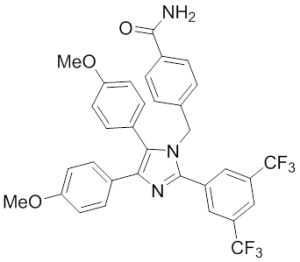
Methylene blue
Methylene blue (MB) was identified as an inhibitor of the ATPase activity of Hsp70-1 266. MB irreversibly inactivates Hsp70-1 but not Hsp70-8 (Hsc70, HSPA8) by oxidizing Cys306 which is not conserved in Hsp70-8 270. It reduces the levels of several Hsp70-1 substrates, including Tau, polyglutamine fragments and Akt, in vitro an in vivo 157, 266, 271, 272. MB also improves cognitive functions in mouse models of Alzheimer’s disease (AD) 271, 273 and has been explored in Phase IIb clinical trials in AD patients 274. Due to its enviable safety record, MB is used clinically for multiple indications 275.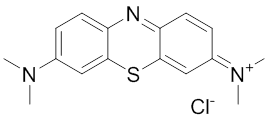
Peptides
As mentioned before, DnaK is considered as being a potential target in antibacterial therapies. Pharmacological inhibitors including drosocin, pyrrhocoricin, and apidaecin have been reported to bind to DnaK and selectively kill susceptible bacteria 276. Pyrrhocoricin binds to the SBD of E. coli DnaK thereby inhibiting its ATPase activity 277, but it does not interact with S. aureus DnaK 278. The generation of synthetic analogs such as the proline-rich antibacterial peptide dimer A3-APO improved stability and activity of the agent 279. Remarkably, A3-APO elicits cytotoxicity against E. coli and Enterobactericae spec. without affecting eukaryotic cells. In mouse models of S. aureus and Propionibacterium acnes intradermal infections, A3-APO and its monomeric metabolite very efficiently ameliorated resistant aerobic and anaerobic intradermal infections 280. A3-APO has recently been reported to reduce the amount of properly folded Bacillus cereus diarrhoeal enterotoxin production, to restrict the proliferation of lethal toxin-induced B. anthracis replication in mouse macrophages, and to improve survival in systemic mouse challenge models 281. According to Otvos et al., the inhibition of bacterial protein folding in synergy with other types of antimicrobial modes might represent a novel strategy in combating resistant or life-threatening infections 281 even though these compounds have not entered clinical trials up to date.
Phenylethylsulfonamide
The small molecule 2-phenylethynesulfonamide (PES) was recently identified as a novel Hsp70-1 inhibitor 88. This compound alters co-chaperone association with Hsp70-1 and impairs the autophagal/lysosomal system as well as the proteasomal pathway consequently leading to the functional inactivity of several cellular proteins including HSP70s and client proteins and, through this activity, impacting substrate fate. Balaburski and colleagues found out that PES binds to the SBD of Hsp70-1 thereby requiring the C-terminal helical “lid” of this protein for binding 282. In animal models of spontaneous B cell lymphoma, administration of PES or the related compound 2-(3-chlorophenyl)ethynesulfonamide (PES-Cl), which shows increased cytotoxicity and ability to inhibit autophagy, significantly protected mice from B cell lymphoma development without any sign of organ toxicity 88, 282. These findings extend previous observations on the cytotoxic activity of PES against leukemia cell lines 283, 284. French et al. demonstrated that Hsp70, Hsp90, and several co-chaperones functionally co-localize with a dynamic multiprotein complex involved in purine synthesis, called the purinosome 285. Treatment of HeLa cells with PES produced a reversible disruption of the purinosome. The combination of PES with the known cancer drug methotrexate synergistically increased cytotoxic effects on cervical cancer cells 285. Since the direct inhibition of HSP70s (Hsp70-1 and Hsp70-8) by PES and VER-155008 induced an early elongation pausing of ribosomes and concomitant downregulation of protein translation, PES and its derivatives can be considered as an auspicious group of HSP70 inhibitors holding promise for innovative anticancer strategies.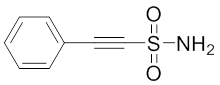
Rhodocyanines and derivatives
The rhodocyanine MKT-077 preferentially binds and inhibits the ADP-bound form of HSP70s such as Hsp70-9 (Grp75) and Hsp70-8 (Hsc70) 286, 287. This allosteric HSP70 inhibitor has originally been described to exhibit antitumor activity in preclinical studies based on selective mitochondrial accumulation 288. Although the antitumor activity of MKT-077 was confirmed using human tumor xenografts in nude mice 289, a phase I trial in patients with chemoresistant advanced solid cancers revealed functional renal impairment necessitating early termination of the study 290. Noteworthy, combined inhibition of Hsp90 and Hsp70-5 (Grp75) with 17-AGG and MKT-077 was recently found to confer potent antitumor effects against hepatocellular carcinomas through blockage of Akt activation and p53 accumulation in vivo, providing an additional therapeutic strategy for liver cancer therapy 291.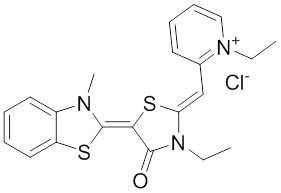
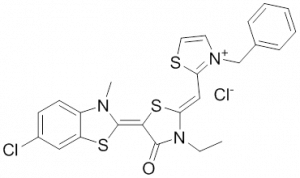
Meanwhile it became apparent that MKT-077 is rapidly metabolized, which limits its use as either a chemical probe or potential therapeutic leading to the synthesis of analogs with greater stability.
The optimization MKT-077 scaffold led to the identification of new compounds with improved drug-like properties. Among them, JG-98 was found as being at least 3-fold more active than MKT-077 against the breast cancer cell lines MDA-MB-231 and MCF-7 (EC50 values of 400 nM) 292. JG-98 has been shown to destabilize the chaperone client proteins such as Akt and Raf-1, and induce apoptosis in these cells. A further microsomal stable MKT-007 analog, JG-13 has been reported as being an active anticancer compound, with IC50 values in the lower µM range against MCF-7 and MDA-MB-231 cells 292. JG-13 interferes with Hsp70-8/Bag-1 interactions in vitro and reduces both, phosphorylated and total Tau in Tau-overexpressing HeLa cells. JG-13 also rescued long-term potentiation deficits in hippocampal sclices from rTg4510 mice. Similarly, JG-48 suppresses phosphorylated and total Tau in Tau-overexpressing HeLa cells, endogenous Tau in neuroblastoma cells, and endogenous Tau in primary neurons from rTg4510 mice, a mouse model of Alzheimer’s disease 293.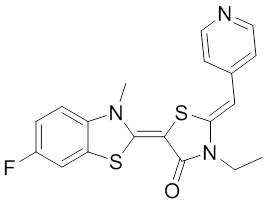
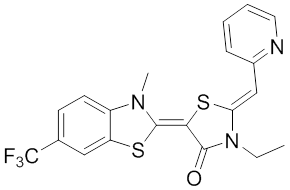
A novel class of HSP70 inhibitors is represented by the stable and soluble MKT-077 analog YM-01 harboring promising cytotoxic activities in certain cancer cell lines without affecting normal or immortalized cells 294.
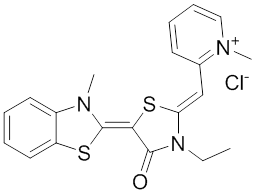
In the same study, treatment with YM-01 also restored tamoxifen sensitivity to a refractory tamoxifen-resistant MCF7 cell model implying that YM-01 holds great potential as a cancer therapeutic. Recent data by the group of Chad Dickey convincingly revealed that YM-01 reduces Tau levels in Tau-overexpressing HeLa cells, endogenous Tau in neuroblastoma cells, and endogenous Tau in primary neurons from the neurodegenerative mouse model rTg4510 293. YM-01 also rescued long-term potentiation deficits in hippocampal slices from rTg4510 mice.
YM-08, a neutral analog of MKT-077 able to penetrate the blood-brain barrier, binds to Hsp70-8 in vitro and reduces phosphorylated Tau levels in cultured brain slices identifying YM-08 as a promising scaffold for the development of HSP70 inhibitors suitable for use in the CNS 295.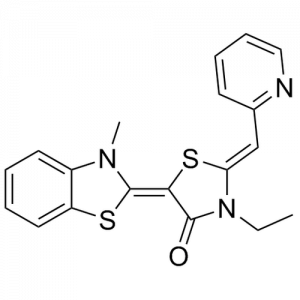
Sulfoglycolipids
Sulfogalactoglycerolipid and sulfogalactosylceramide (SGC) are two sulfoglycolipids capable of binding to Hsp70 296, 297. Substitution of the acyl chain by an adamantly group (adaSGC) improved Hsp70 affinity 297. In transfected baby hamster kidney cells, adaSGC increased protein levels of DF508 CFTR, a mutant of the cystis fibrosis transmembrane receptor (CFTR) that tend to misfolding and decomposition via ER-mediated degradation 298. From these results it can be concluded that blockage of the ATPase activity of HSP70s might repress ER-mediated degradation pathways that physiologically reduces the amount of mutated CFTR 255. These observations might have future clinical implications with respect to the use of HSP70s as drug targets in neurodegenerative misfolding disorders.